Single-cell tutorial
This tutorial provides a step by step introduction on how to run nf-core/airrflow on single-cell BCR-seq data or single-cell TCR-seq data.
Pre-requisites
If you are new to Nextflow and nf-core, please refer to this page on how to set up Nextflow and a container engine needed to run this pipeline. At the moment, nf-core/airrflow does NOT support using conda virtual environments for dependency management, only containers are supported. Make sure to test your setup before running the workflow on actual data.
For the purpose of running this tutorial on your local machine, we recommend a Docker installation. To install Docker, follow the instructions here. After Docker installation on Linux system, don’t forget to check the post-installation steps.
Alternatively, you can run this tutorial using the Gitpod platform which contains pre-installed Nextflow, nf-core and Docker. There are three ways to open Gitpod platform. Please watch this video to set it up. If you want to know more about Gitpod, check the Gitpod overview.
Testing the pipeline with built-in tests
Once you have set up your Nextflow and container (Docker or Singularity), test nf-core/airrflow with the built-in test data.
nextflow run nf-core/airrflow -r 4.3.0 -profile test,docker --outdir test_resultsIf the tests run through correctly, you should see the execution of airrflow processes. Finally, the following output will appear in your command line:
output:
-[nf-core/airrflow] Pipeline completed successfully-
Completed at: 11-Mar-2025 11:30:35
Duration : 5m 50s
CPU hours : 0.6
Succeeded : 221Supported input formats
There are two supported input formats for nf-core/airrflow single-cell AIRR-seq pipeline: assembled sequences in AIRR rearrangement format or raw reads in fastq format sequenced in the 10x Genomics platform.
The AIRR rearrangement format is a standard format to store BCR and TCR sequence data with relevant metadata fields. This format is supported as input and output by multiple tools specific for analyzing AIRR-seq data. For example, when analyzing single-cell AIRR sequencing data with CellRanger versions >= 4.0 an AIRR rearrangement file will be provided as output, and this is the recommended input for running nf-core/airrflow. Note that it is also possible to start running the pipeline directly from raw sequencing reads, and in this case CellRanger will be run when launching nf-core/airrflow.
The AIRR rearrangement format is also the default one when analyzing publicly available data from specialized AIRR-seq databases such as the AIRR Data Commons through the iReceptor gateway.
In this tutorial we will showcase how to run nf-core/airrflow with both of the input formats.
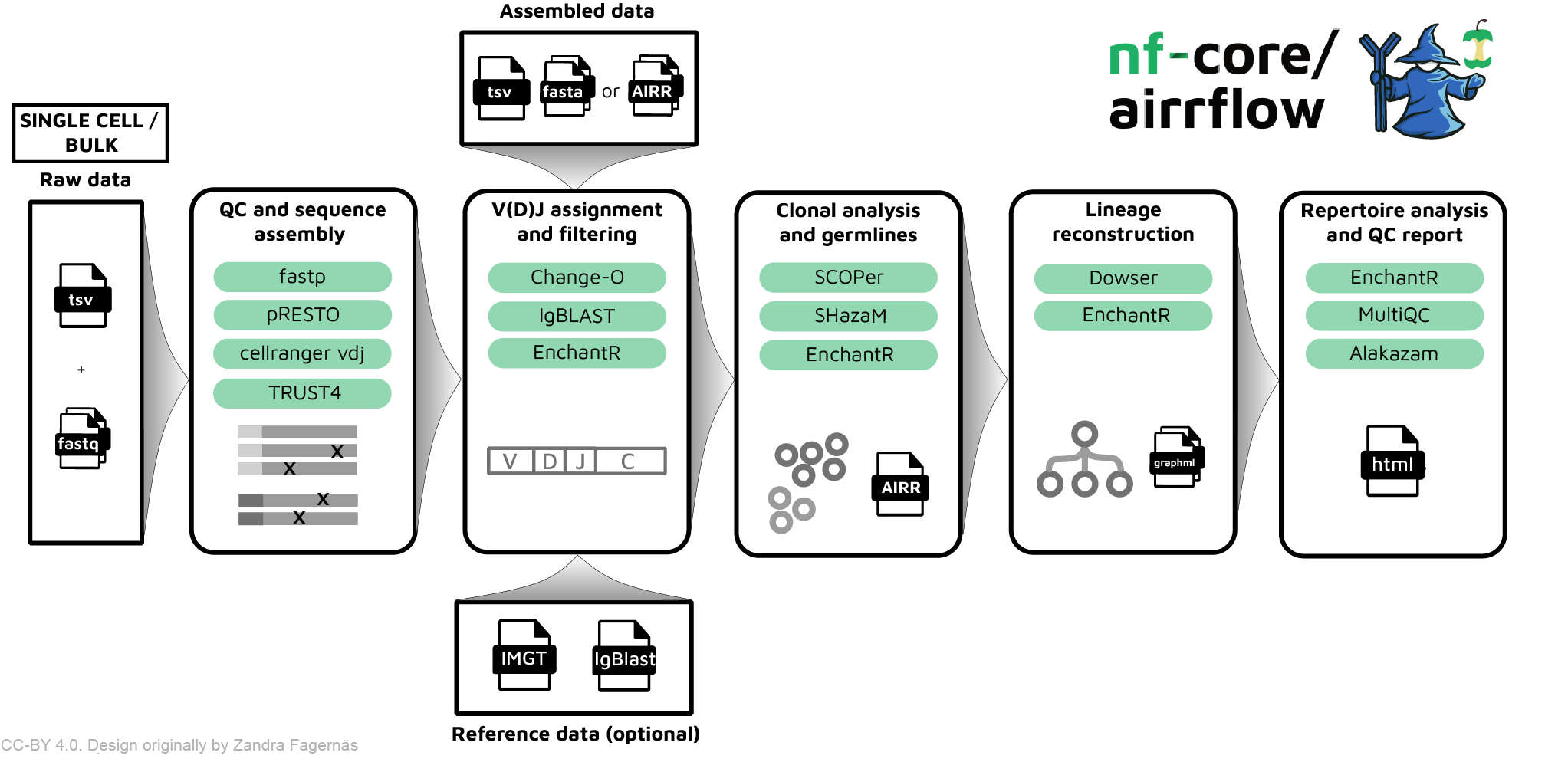
Starting from AIRR rearrangement format
Datasets
For this tutorial we will use sub-sampled PBMC single-cell BCR sequencing data from two subjects, before (d0) and after flu vaccination (d12). The dataset is publicly available on Zenodo. You don’t need to download the dataset because the links to the samples are already provided in the samplesheet and Nextflow will get the data from the links automatically when running the pipeline.
Preparing the samplesheet and configuration file
To run the pipeline, a tab-separated samplesheet that provides the path to the AIRR rearrangement files must be prepared. The samplesheet collects experimental details that are important for the data analysis.
Details on the required columns of a samplesheet are available here.
The resource configuration file sets the compute infrastructure maximum available number of CPUs, RAM memory and running time. This will ensure that no pipeline process requests more resources than available in the compute infrastructure where the pipeline is running. The resource config should be provided with the -c option. In this example we set the maximum RAM memory to 16GB, we restrict the pipeline to use 8 CPUs and to run for a maximum of 24 hours.
process {
resourceLimits = [ memory: 16.GB, time: 24.h, cpus: 8 ]
}A prepared samplesheet for this tutorial can be found here, and the configuration file is available here. Download both files to the directory where you intend to run nf-core/airrflow.
Before setting memory and cpus in the configuration file, we recommend verifying the available memory and cpus on your system. Otherwise, exceeding the system’s capacity may result in an error indicating that you requested more cpus than available or run out of memory. You can also remove the “time” parameter from the configuration file to allow for unlimited runtime for large-size dataset.
When running nf-core/airrflow with your own data, provide the full path to your input files under the filename column.
Running airrflow
With all the files ready, you can proceed to start the pipeline run:
nextflow run nf-core/airrflow -r 4.3.0 \
-profile docker \
--mode assembled \
--input assembled_samplesheet.tsv \
--outdir sc_from_assembled_results \
-c resource.config \
-resumeOf course you can wrap all your code in a bash file. We prepared one for you and it’s available here. With the bash file, it’s easy to run the pipeline with a single-line command.
bash airrflow_sc_from_assembled.shWhen launching a Nextflow pipeline with the -resume option, any processes that have already been run with the exact same code, settings and inputs will be cached and the pipeline will resume from the last step that changed or failed with an error. The benefit of using “resume” is to avoid duplicating previous work and save time when re-running a pipeline.
We include “resume” in our Nextflow command as a precaution in case anything goes wrong during execution. After fixing the issue, you can relaunch the pipeline with the same command, it will resume running from the point of failure, significantly reducing runtime and resource usage.
After launching the pipeline the following will be printed to the console output, followed by some the default parameters used by the pipeline and execution log of airrflow processes:
N E X T F L O W ~ version 24.10.5
WARN: It appears you have never run this project before -- Option `-resume` is ignored
Launching `https://github.com/nf-core/airrflow` [boring_heyrovsky] DSL2 - revision: d91dd840f4 [4.3.0]
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/airrflow 4.3.0
------------------------------------------------------
Once the pipeline has finished successfully, the following message will appear:
-[nf-core/airrflow] Pipeline completed successfully-
Completed at: 11-Mar-2025 13:06:05
Duration : 2m 47s
CPU hours : 0.4
Succeeded : 44Starting from raw reads in fastq format
Datasets
For this tutorial we will use subsampled blood single-cell TCR sequencing data of one subject generated from the 10x Genomic platform. The links to the fastq files are in the samplesheet.
Preparing samplesheet, gene reference and configuration file
To run nf-core/airrflow on single cell TCR or BCR sequencing data from fastq files, we need to prepare samplesheet, pre-built 10x genomics V(D)J references and configuration file in advance. Details on the required columns for this samplesheet are available here.
The fastq file names must follow the 10X Genomics file naming convention or the cellranger process will fail.
The prepared samplesheet for this tutorial is here and a prepared configuration file is here. Download these two files to the directory where you intend to run nf-core/airrflow.
Before setting memory and cpus in the configuration file, we recommend verifying the available memory and cpus on your system. Otherwise, exceeding the system’s capacity may result in an error indicating that you requested more cpus than available or run out of memory.
Pre-built 10x genomics V(D)J references can be accessed at the 10x Genomics website. Both human and mouse V(D)J references are available. Download the reference that corresponds to the species of your dataset.
Running airrflow
With all the files ready, it’s time to run nf-core/airrflow.
nextflow run nf-core/airrflow -r 4.3.0 \
-profile docker \
--mode fastq \
--input 10x_sc_raw.tsv \
--library_generation_method sc_10x_genomics \
--reference_10x refdata-cellranger-vdj-GRCh38-alts-ensembl-7.1.0 \
-c resource.config \
--clonal_threshold 0 \
--outdir sc_from_fastq_results \
-resumeIn this tutorial, since the samples are TCRs, which do not have somatic hypermutation, clones are defined strictly by identical junction regions. For this reason, we set the --clonal_threshold parameter to 0. For more details on important considerations when performing clonal analysis check FAQ.
Of course you can wrap all your code in a bash file. We prepared one for you and it’s available here. With the bash file, it’s easy to run the pipeline with a single-line command.
bash airrflow_sc_from_fastq.shAfter launching the pipeline the following will be printed to the console output, followed by some the default parameters used by the pipeline and execution log of airrflow processes:
N E X T F L O W ~ version 24.10.5
Launching `https://github.com/nf-core/airrflow` [gloomy_monod] DSL2 - revision: d91dd840f4 [4.3.0]
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/airrflow 4.3.0
------------------------------------------------------Once the pipeline has finished successfully, the following message will appear:
-[nf-core/airrflow] Pipeline completed successfully-
Completed at: 11-Mar-2025 13:18:13
Duration : 2m 46s
CPU hours : 0.3 (0.1% cached)
Succeeded : 17
Cached : 2Understanding the results
After running the pipeline, several sub-folders are available under the results folder.
Airrflow_report.html
- cellranger
- vdj_annotation
- qc_filtering
- clonal_analysis
- repertoire_comparison
- multiqc
- report_file_size
- pipeline_infoThe summary report, named Airrflow_report.html, provides an overview of the analysis results, such as an overview of the number of sequences per sample in each of the pipeline steps, the V(D)J gene assignment and QC, and V gene family usage. Additionally, it contains links to detailed reports for other specific analysis steps.
The analysis steps and their corresponding folders, where the results are stored, are briefly listed below. Detailed documentation on the pipeline output can be found on the Output documentation page.
-
QC and sequence assembly (if starting from fastq files).
- In this first step, Cell Ranger’s VDJ algorithm is employed to assemble contigs, annotate contigs, call cells and generate clonoytpes. The results are stored in the ‘cellranger’ folder.
-
V(D)J annotation and filtering.
- In this step, V(D)J gene segments are inferred using the provided germline reference and
IgBLAST. Alignments are annotated in AIRR format. Non-productive sequences and sequences with low alignment quality are filtered out unless otherwise specified. The intermediate results are stored under the folder named ‘vdj_annotation’.
- In this step, V(D)J gene segments are inferred using the provided germline reference and
-
QC filtering.
- In this step, cells without heavy chains or with multiple heavy chains are removed. Sequences in different samples that share the same cell_id and nucleotide sequence are filtered out. The result are stored in the ‘qc-filtering’ folder.
-
Clonal analysis.
- Results of the clonal threshold determination using
SHazaMshould be inspected in the html report under the ‘clonal_analysis/find_threshold’ folder. If the automatic threshold is unsatisfactory, you can set the threshold manually and re-run the pipeline. (Tip: use -resume whenever running the Nextflow pipeline to avoid duplicating previous work). - Clonal inference is performed with
SCOPer. Clonal inference results as well as clonal abundance and diversity plots can be inspected in the html report in the folder ‘clonal_analysis/define_clones’. For BCR sequencing data, mutation frequency is also computed usingSHazaMat this step and plotted in the report. Therepertoiressubfolder contains the AIRR formatted files with the clonal assignments in a new columnclone_idand mutation frequency in the columnmu_freq. Thetablessubfolder contains the tabulated abundance and diversity computation as well as a table with the number of clones and their size. Theggplotssubfolder contains the abundance and diversity plots as anRDataobject for loading and customization in R. - If lineage trees were computed using
Dowser, a folder under ‘clonal_analysis/dowser_lineages’ will be present. The trees can be inspected in the html report and saved as PDF. Additionally, anRDSobject with the formatted trees can also be loaded in R for customizing the lineage tree plots with Dowser.
- Results of the clonal threshold determination using
-
Repertoire analysis
- Comparison of several repertoire characteristics, such as V gene usage, across subjects, time points or cell populations. All associated plots and tables are available under the folder
repertoire_comparison. The plots are also included in theAirrflow_report.htmlfile. This report is generated from an R markdownRmdfile. It is possible to customize this to meet the user’s needs by editing the report and then providing the edited Rmd file with the--report_rmdparameter. Check the remaining Report parameters for further customizing the report.
- Comparison of several repertoire characteristics, such as V gene usage, across subjects, time points or cell populations. All associated plots and tables are available under the folder
-
Other reporting. Additional reports are also generated, including:
- MultiQC report: summarizes QC metrics across all samples.
- Pipeline_info report: various reports relevant to the running and execution of the pipeline.
- Report_file_size report: Summary of the number of sequences left after each of the most important pipeline steps.
Find out more
To continue learning about how to use nf-core/airrflow please check out the following documentation:
The nf-core troubleshooting documentation will also help you troubleshoot your Nextflow errors